
Hi! I'm Colby...
-

Mathematics
Statistical Modeling
Bayesian Nets
Operations Research
Numerical Logic -

Machine Learning
Algorithm Design
Natural Language Processing
Deep Learning
Artificial Intelligence -

Computational Biology
Human Genomics
Phylogenetics
Protein Modeling
Infectious Diseases -

Distributed Computing
Apache Spark (Databricks)
Cluster Computing (MPI/SNOW)
GPU-Based Processing (CUDA)
High Performance Computing -

Programming
R/SparkR
Python/PySpark
SQL
Visual Basic -

Web and Design
HTML5+CSS
LaTeX
API Development
Visualization
-
Genomics, Machine Learning, Influenza
Avian Influenza Modeling of Antibody Binding Affinity
Large-Scale Computational Modeling of H5 Influenza Variants Against HA1-Neutralizing Antibodies
The United States Department of Agriculture has recently released reports that show samples from 2022-2024 of highly pathogenic avian influenza (H5N1) have been detected in mammals and birds (1). To date, the United States Centers for Disease Control reports that there have been 27 humans infected with H5N1 in 2024 (2). The broader potential impact on human health remains unclear. In this study, we computationally model 1,804 protein complexes consisting of various H5 isolates from 1959 to 2024 against 11 hemagglutinin domain 1 (HA1)-neutralizing antibodies. This study shows a trend of weakening binding affinity of existing antibodies against H5 isolates over time, indicating that the H5N1 virus is evolving immune escape of our medical defenses. We also found that based on the wide variety of host species and geographic locations in which H5N1 was observed to have been transmitted from birds to mammals, there is not a single central reservoir host species or location associated with H5N1’s spread. These results indicate that the virus has potential to move from epidemic to pandemic status in the near future. This study illustrates the value of high-performance computing to rapidly model protein-protein interactions and viral genomic sequence data at-scale for functional insights into medical preparedness.
-
Genomics, Machine Learning, Oncology
AI-Based Antibody Diffusion Targeting PD-1 in Oncology
PD-1 Targeted Antibody Discovery Using AI Protein Diffusion
Understanding the interactions between SARS-CoV-2 and the human immune system is paramount to the characterization of novel variants as the virus co-evolves with the human host. In this study, we employed state-of-the-art molecular docking tools to conduct large-scale virtual screens, predicting the binding affinities between 64 human cytokines against 17 nucleocapsid proteins from six betacoronaviruses. Our comprehensive in silico analyses reveal specific changes in cytokine-nucleocapsid protein interactions, shedding light on potential modulators of the host immune response during infection. These findings offer valuable insights into the molecular mechanisms underlying viral pathogenesis and may guide the future development of targeted interventions. This manuscript serves as insight into the comparison of deep learning based AlphaFold2-Multimer and the semi-physicochemical based HADDOCK for protein-protein docking. We show the two methods are complementary in their predictive capabilities. We also introduce a novel algorithm for rapidly assessing the binding interface of protein-protein docks using graph edit distance: graph-based interface residue assessment function (GIRAF). The high-performance computational framework presented here will not only aid in accelerating the discovery of effective interventions against emerging viral threats, but extend to other applications of high throughput protein-protein screens.
-
Genomics, Machine Learning, SARS-CoV-2
SARS-CoV-2 BA.2.86 and JN.1 Modeling of Antibody and ACE2 Binding Affinity
Predicting antibody and ACE2 affinity for SARS-CoV-2 BA.2.86 and JN.1 with in silico protein modeling and docking
The emergence of SARS-CoV-2 lineages derived from Omicron, including BA.2.86 (nicknamed “Pirola”) and its relative, JN.1, has raised concerns about their potential impact on public and personal health due to numerous novel mutations. Despite this, predicting their implications based solely on mutation counts proves challenging. Empirical evidence of JN.1’s increased immune evasion capacity in relation to previous variants is mixed. To improve predictions beyond what is possible based solely on mutation counts, we conducted extensive in silico analyses on the binding affinity between the RBD of different SARS-CoV-2 variants (Wuhan-Hu-1, BA.1/B.1.1.529, BA.2, XBB.1.5, BA.2.86, and JN.1) and neutralizing antibodies from vaccinated or infected individuals, as well as the human angiotensin-converting enzyme 2 (ACE2) receptor. We observed no statistically significant difference in binding affinity between BA.2.86 or JN.1 and other variants. Therefore, we conclude that the new SARS-CoV-2 variants have no pronounced immune escape or infection capacity compared to previous variants. However, minor reductions in binding affinity for both the antibodies and ACE2 were noted for JN.1. Future research in this area will benefit from increased structural analyses of memory B-cell derived antibodies and should emphasize the importance of choosing appropriate samples for in silico studies to assess protection provided by vaccination and infection. Moreover, the fitness benefits of genomic variation outside of the RBD of BA.2.86 and JN.1 need to be investigated. This research contributes to understanding the BA.2.86 and JN.1 variants’ potential impact on public health.
-
Genomics, RNA-seq, Zebrafish
mecp2 function in zebrafish
MeCP2's function as a transcriptional regulator is conserved across vertebrates and supports using zebrafish to complement mouse modeling in elucidating these conserved mechanisms.
Rett syndrome (RTT), a human neurodevelopmental disorder characterized by severe cognitive and motor impairments, is caused by dysfunction of the conserved transcriptional regulator Methyl-CpG-binding protein 2 (MECP2). Genetic analyses in mouse Mecp2 mutants, which exhibit key features of human RTT, have been essential for deciphering the mechanisms of MeCP2 function; nonetheless, our understanding of these complex mechanisms is incomplete. Zebrafish mecp2 mutants exhibit mild behavioral deficits but have not been analyzed in depth. Here, we combine transcriptomic and behavioral assays to assess baseline and stimulus-evoked motor responses and sensory filtering in zebrafish mecp2 mutants from 5 to 7 days post-fertilization (dpf). We show that zebrafish mecp2 function is required for normal thigmotaxis but is dispensable for gross movement, acoustic startle response, and sensory filtering (habituation and sensorimotor gating), and reveal a previously unknown role for mecp2 in behavioral responses to visual stimuli. RNA-seq analysis identified a large gene set that requires mecp2 function for correct transcription at 4 dpf, and pathway analysis revealed several pathways that require MeCP2 function in both zebrafish and mammals. These findings show that MeCP2's function as a transcriptional regulator is conserved across vertebrates and supports using zebrafish to complement mouse modeling in elucidating these conserved mechanisms.
-
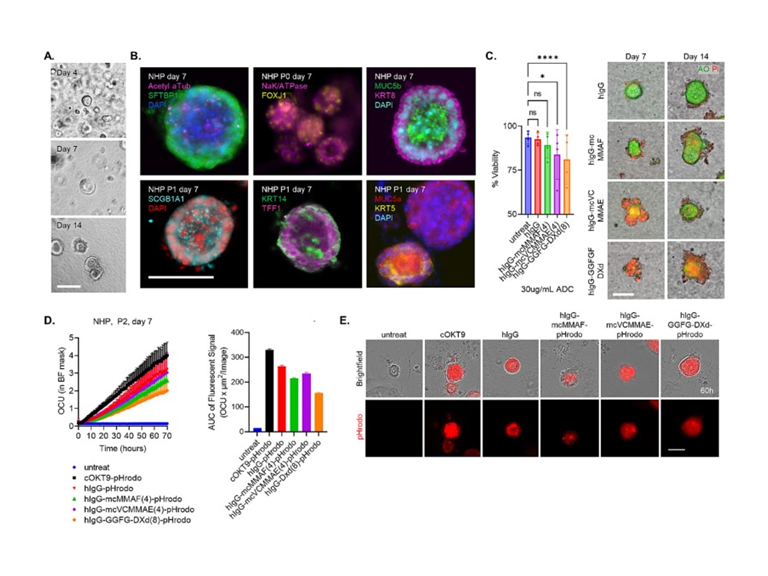
Genomics, Oncology, Antibodies
Bronchioalveolar organoids in ADC toxicity
Bronchioalveolar organoids: A preclinical tool to screen toxicity associated with antibody-drug conjugates
Despite extensive preclinical testing, cancer therapeutics can result in unanticipated toxicity to non-tumor tissue in patients. These toxicities may pass undetected in preclinical experiments due to modeling limitations involving poor biomimicry of 2-dimensional in vitro cell cultures and due to lack of interspecies translatability in in vivo studies. Instead, primary cells can be grown into miniature 3-dimensional structures that recapitulate morphological and functional aspects of native tissue, termed “organoids.” Here, human bronchioalveolar organoids grown from primary alveolar epithelial cells were employed to model lung epithelium and investigate off-target toxicities associated with antibody-drug conjugates (ADCs). ADCs with three different linker-payload combinations (mafodotin, vedotin, and deruxtecan) were tested in bronchioalveolar organoids generated from human, rat, and nonhuman primate lung cells. Organoids demonstrated antibody uptake and changes in viability in response to ADC exposure that model in vivo drug sensitivity. RNA sequencing identified inflammatory activation in bronchioalveolar cells in response to deruxtecan. Future studies will explore specific cell populations involved in interstitial lung disease and incorporate immune cells to the culture.
-
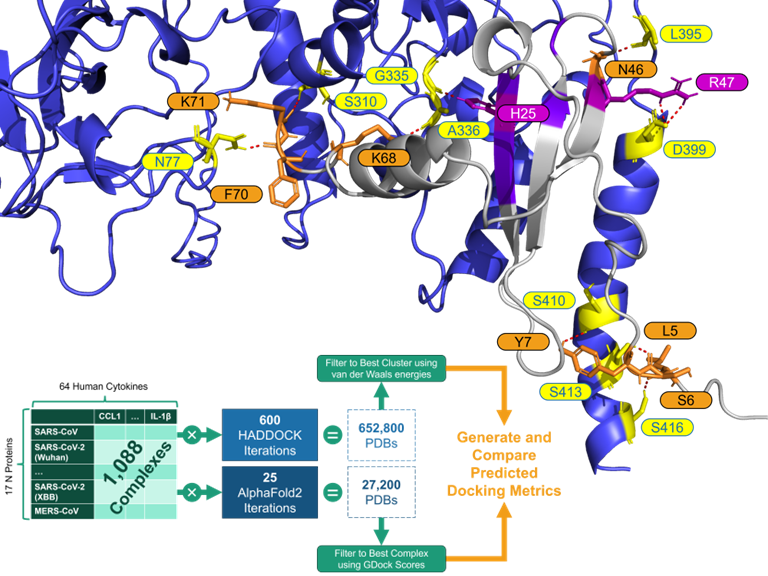
Genomics, Machine Learning, SARS-CoV-2
Human Cytokine and SARS-CoV-2 N interactivity
Human Cytokine and Coronavirus Nucleocapsid Protein Interactivity Using Large-Scale Virtual Screens
Understanding the interactions between SARS-CoV-2 and the human immune system is paramount to the characterization of novel variants as the virus co-evolves with the human host. In this study, we employed state-of-the-art molecular docking tools to conduct large-scale virtual screens, predicting the binding affinities between 64 human cytokines against 17 nucleocapsid proteins from six betacoronaviruses. Our comprehensive in silico analyses reveal specific changes in cytokine-nucleocapsid protein interactions, shedding light on potential modulators of the host immune response during infection. These findings offer valuable insights into the molecular mechanisms underlying viral pathogenesis and may guide the future development of targeted interventions. This manuscript serves as insight into the comparison of deep learning based AlphaFold2-Multimer and the semi-physicochemical based HADDOCK for protein-protein docking. We show the two methods are complementary in their predictive capabilities. We also introduce a novel algorithm for rapidly assessing the binding interface of protein-protein docks using graph edit distance: graph-based interface residue assessment function (GIRAF). The high-performance computational framework presented here will not only aid in accelerating the discovery of effective interventions against emerging viral threats, but extend to other applications of high throughput protein-protein screens.
-
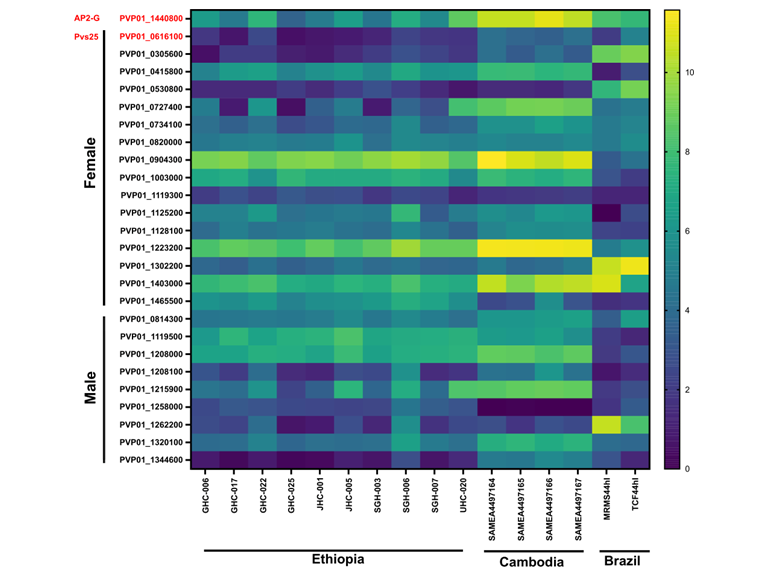
Genomics, scRNA-seq, Plasmodium
Comparative transcriptomics of Plasmodium vivax
Comparative transcriptomics reveal differential gene expression among Plasmodium vivax geographical isolates and implications on erythrocyte invasion mechanisms
The documentation of Plasmodium vivax malaria across Africa, especially in regions where Duffy negatives are dominant, suggests possibly alternative erythrocyte invasion mechanisms. While the transcriptomes of the Southeast Asian and South American P. vivax are well documented, the gene expression profile of P. vivax in Africa is unclear. In this study, we examined the expression of 4,404 gene transcripts belong to 12 functional groups and 43 erythrocyte binding gene candidates in Ethiopian isolates and compared them with the Cambodian and Brazilian P. vivax transcriptomes. Overall, there were 10–26% differences in the gene expression profile amongst geographical isolates, with the Ethiopian and Cambodian P. vivax being most similar. Majority of the gene transcripts involved in protein transportation, housekeeping, and host interaction were highly transcribed in the Ethiopian isolates. Members of the reticulocyte binding protein PvRBP2a and PvRBP3 expressed six-fold higher than Duffy binding protein PvDBP1 and 60-fold higher than PvEBP/DBP2 in the Ethiopian isolates. Other genes including PvMSP3.8, PvMSP3.9, PvTRAG2, PvTRAG14, and PvTRAG22 also showed relatively high expression. Differential expression patterns were observed among geographical isolates, e.g., PvDBP1 and PvEBP/DBP2 were highly expressed in the Cambodian but not the Brazilian and Ethiopian isolates, whereas PvRBP2a and PvRBP2b showed higher expression in the Ethiopian and Cambodian than the Brazilian isolates. Compared to Pvs25, gametocyte genes including PvAP2-G, PvGAP (female gametocytes), and Pvs47 (male gametocytes) were highly expressed across geographical samples.
-
Genomics, Phylogenetics, Echinodermata
Occurrence of the starfish Luidia magnifica in the Mexican Pacific
Occurrence of the Indo-West Pacific starfish Luidia magnifica (Echinodermata: Asteroidea) in the Mexican Pacific and a possible introduction to the Caribbean region
The starfish Luidia magnifica (Asteroidea: Paxillosida: Luidiidae) is a multiradiate starfish that occurs throughout the Indo-Pacific. Since the description of L. magnifica in 1906 by Fisher, very few observations of this species have been documented. We present new shallow-water records for L. magnifica in the Tropical Eastern Pacific obtained between 2017 and 2021. We collected a total of 15 specimens of L. magnifica in the bay of Manzanillo, Colima, in the central nearshore Mexican Pacific. We provide morphological description of specimens collected and additional ecological data. We used molecular techniques to verify species identification. This record represents a considerable expansion of the known distribution range of L. magnifica and provides the first evidence of the presence of this species in the Tropical Eastern Pacific. The presence of L. magnifica larvae from the Caribbean coast of Panama was also identified from DNA sequencing of larvae left unindentified in previous reports. This result indicates that it is possible that the L. magnifica also occurs in the Carribbean. Because observations of the starfish in the Mexican Pacific have been recent, the presence of larvae in the Caribbean could be a recent introduction through the Panama Canal from the Eastern Pacific to the Western Atlantic.
-
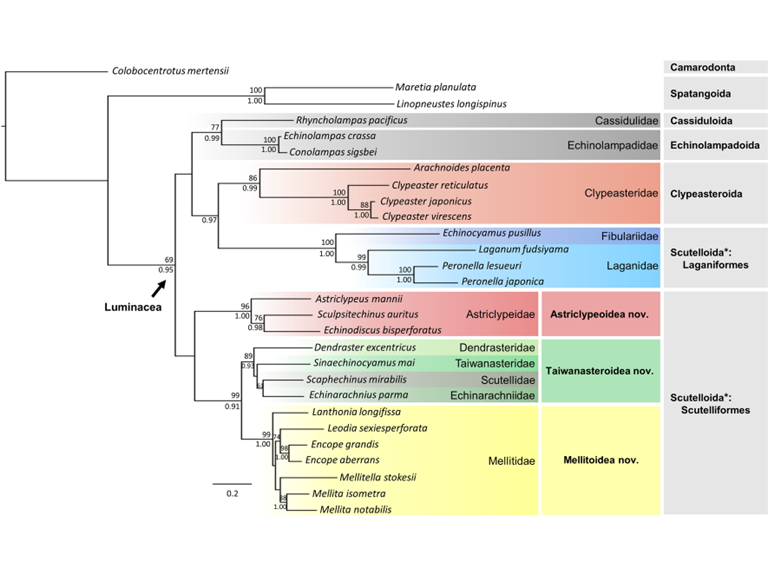
Genomics, Phylogenetics, Echinodermata
Phylogeny, ancestral ranges and reclassification of sand dollars
Phylogeny, ancestral ranges and reclassification of sand dollars
Classification of the Class Echinoidea is under significant revision in light of emerging molecular phylogenetic evidence. In particular, the sister-group relationships within the superorder Luminacea (Echinoidea: Irregularia) have been considerably updated. However, the placement of many families remains largely unresolved due to a series of incongruent evidence obtained from morphological, paleontological, and genetic data for the majority of extant representatives. In this study, we investigated the phylogenetic relationships of 25 taxa, belonging to eleven luminacean families. We proposed three new superfamilies: Astriclypeoidea, Mellitoidea, and Taiwanasteroidea (including Dendrasteridae, Taiwanasteridae, Scutellidae, and Echinarachniidae), instead of the currently recognized superfamily Scutelloidea Gray, 1825. In light of the new data obtained from ten additional species, the historical biogeography reconstructed shows that the tropical western Pacific and eastern Indian Oceans are the cradle for early sand dollar diversification. Hothouse conditions during the late Cretaceous and early Paleogene were coupled with diversification events of major clades of sand dollars. We also demonstrate that Taiwan fauna can play a key role in terms of understanding the major Cenozoic migration and dispersal events in the evolutionary history of Luminacea.
-
Genomics, SARS-CoV-2, Machine Learning
SARS-CoV-2 XBB.1.5 RBD-Antibody Predictions
Predicting changes in neutralizing antibody activity for SARS-CoV-2 XBB.1.5 using in silico protein modeling..
The SARS-CoV-2 variant XBB.1.5 is of concern as it has high transmissibility. XBB.1.5 currently accounts for upwards of 30% of new infections in the United States. One year after our group published the predicted structure of the Omicron (B.1.1.529) variant's receptor binding domain and antibody binding affinity, we return to investigate the new mutations seen in XBB.1.5 which is a descendant of Omicron. Using in silico modeling approaches against newer neutralizing antibodies that are shown effective against B.1.1.529, we posit the immune consequences of XBB.1.5's mutations and show that there is no statistically significant difference in overall antibody evasion when comparing to the B.1.1.529 and other related variants (e.g. BJ.1 and BM.1.1.1). However, noticeable changes in neutralizing activity were seen due to specific amino acid changes of interest in the newer variants.
-

Genomics, Cloud, Machine Learning
Genomics in the Azure Cloud Book
Building a cloud genomics architecture in Azure using enterprise-grade platform services.
This practical guide bridges the gap between general cloud computing architecture in Microsoft Azure and scientific computing for bioinformatics and genomics. You'll get a solid understanding of the architecture patterns and services that are offered in Azure and how they might be used in your bioinformatics practice. You'll get code examples that you can reuse for your specific needs. And you'll get plenty of concrete examples to illustrate how a given service is used in a bioinformatics context.
-

Genomics, SARS-CoV-2, Machine Learning
SARS-CoV-2 Omicron RBD Predictions
Deep learning-based predictions of the SARS-CoV-2 Omicron (B.1.1.529) variant receptor binding domain with neutralizing antibodies.
The genome of the SARS-CoV-2 Omicron variant (B.1.1.529) was released on November 22, 2021, which has caused a flurry of media attention due the large number of mutations it contains. These raw data have spurred questions around vaccine efficacy. Given that neither the structural information nor the experimentally-derived antibody interaction of this variant are available, we have turned to predictive computational methods to model the mutated structure of the spike protein's receptor binding domain and posit potential changes to vaccine efficacy. In this study, we predict some structural changes in the receptor-binding domain that may reduce antibody interaction without completely evading existing neutralizing antibodies (and therefore current vaccines).
-

Genomics, Distributed Computing, Data Engineering
Genomics Data Lake eBook
An eBook designed to help bioinformatics teams scale up their genomics research in the Azure cloud.
Centralizing your data in a data lake has the potential to help scale and automate bioinformatics pipelines (including secondary and tertiary analyses and machine learning) in cloud. Azure Data Lake is touted as a limitless service place for storing your data. It provides the ability to store and organize petabyte-size files and connect to distributed computing resources (like Azure Databricks) with ease. In addition, Data Lake offers enterprise-grade security and role-based access controls.
In this book, we discuss the utility of Azure Data Lake and how this flexible and scalable storage option promotes collaboration and scalability in your genomics practice while also ensuring a secure and stable environment for your genomics data. Plus, with its easy integration with other Azure services, orchestrating and automating data movements and bioinformatics pipelines has never been easier (or faster).
-
Genomics, Phylogenetics, Infectious Diseases
P. falciparum CSP Haploytyping
Modeling PfCSP haplotypes and their changes in protein interaction with human immunological proteins.
The world's first malaria vaccine RTS,S provides only partial protection against Plasmodium falciparum infections. The explanation for such low efficacy is unclear. This study examined the associations of parasite genetic variations with binding affinity to human immunological proteins including human leukocyte antigen (HLA) and T cell receptors (TCR) involved in RTS,S-induced immune responses. Multiplicity of infections was determined by amplicon deep sequencing of merozoite surface protein 1 (PfMSP1). Genetic variations in the C-terminal of circumsporozoite protein (PfMSP1) gene were examined across 88 samples of P. falciparum collected from high and low transmission settings of Ghana. Binding interactions of PfMSP1 variants and HLA/TCR were analyzed using NetChop} and HADDOCK predictions. Anti-CSP IgG levels were measured by ELISA in a subset of 10 samples. High polyclonality was detected among P. falciparum infections. A total 27 CSP haplotypes were detected among samples. A significant correlation was detected between the CSP and MSP multiplicity of infection (MOI). No clear clustering of haplotypes was observed by geographic regions. The number of genetic differences in PfCSP between 3D7 and non-3D7 variants does not influence binding interactions to HLA/T cells nor anti-CSP IgG levels. Nevertheless, PfCSP peptide length significantly affects its molecular weight and binding affinity to the HLA. The presence of multiple non-3D7 strains among P. falciparum infections in Ghana impact the effectiveness of RTS,S. Longer PfCSP peptides may elicit a stronger immune response and should be considered in future version RTS,S. The molecular mechanisms of RTS,S cell-mediated immune responses related to longer CSP peptides warrants further investigations.
-
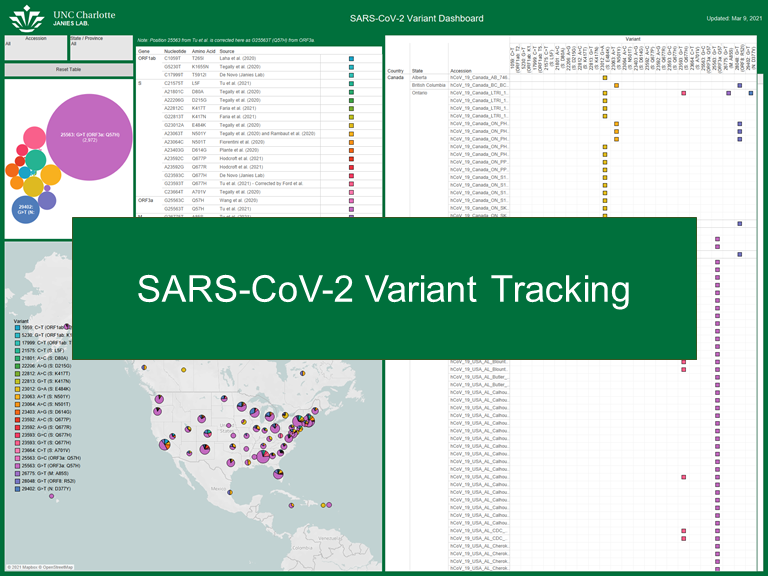
Genomics, Infectious Diseases, Bioinformatics
SARS-CoV-2 Variant Tracking
Building a visual dashboard for cataloging SARS-CoV-2 variants geographically.
Several new variants of the SARS-CoV-2 have been isolated in the United States, Mexico, and Canada. Many of the variants contain single variants of functional significance (e.g. S: N501Y increases transmissibility). To study the occurrence and co-circulation of these variants, we have developed an easy-to-use dashboard.
-

Machine Learning, Distributed Computing, Data Engineering
Sparkitecture
A collection of "cookbook-style" scripts for simplifying data engineering and machine learning in Apache Spark.
Apache Spark is a highly-scalable, massively-parallel computing platform perfect for machine learning and data engineering tasks. Using distributed processing with the Spark API, users can perform various tasks on huge amounts of data using their their preferred language (Python, R, Scala, SQL, etc.), but often there is a bit of a learning curve to using the Spark functionality (PySpark or SparkR) even if the user is a pro at the base language. Sparkitecture is a ebook collection of various script to help make this process a little easier.
-

Genomics, Machine Learning, Infectious Diseases
Machine Learning in Malaria
Machine Learning Modeling in the Prediction of Artemisinin Resistance and Diagnostic Test Sensitivity in Malaria.
Parallel Processing and Ensemble Machine Learning Modeling for the Prediction of Artemisinin Resistance in Malaria (Malaria DREAM Challenge 2019 Submission).
The Malaria DREAM Challenge is open to anyone interested in contributing to the development of computational models that address important problems in advancing the fight against malaria. The overall goal of the first Malaria DREAM Challenge is to predict Artemisinin (Art) drug resistance level of a test set of malaria parasites using their in vitro transcription data and a training set consisting of published in vivo and unpublished in vitro transcriptomes. The in vivo dataset consists of ~1000 transcription samples from various geographic locations covering a wide range of life cycles and resistance levels, with other accompanying data such as patient age, geographic location, Art combination therapy used, etc. [Mok et al., (2015) Science]. The in vitro transcription dataset consists of 55 isolates, with transcription collected at two timepoints (6 and 24 hours post-invasion), in the absence or presence of an Art perturbation, for two biological replicates using a custom microarray at the Ferdig lab. Using these transcription datasets, participants will be asked to predict three different resistance states of a subset of the 55 in vitro isolate samples.
Modeling Plasmodium falciparum Diagnostic Test Sensitivity using Machine Learning with Histidine-Rich Protein 2 Variants.
Malaria, predominantly caused by Plasmodium falciparum, poses one of largest and most durable health threats in the world. Previously, simplistic regression-based models have been created to characterize malaria infections, though these models often only include a couple genetic factors. Specifically, the Baker et al., 2005 model uses two types of particular repeats in histidine-rich protein 2 (PfHRP2) to assert P. falciparum infection, though the efficacy of this model has waned over recent years due to genetic mutations in the parasite. In this work, we use a dataset of 406 P. falciparum PfHRP2 genetic sequences collected in Ethiopia and derived a larger set of motif repeat matches for use in generating a series of diagnostic machine learning models. Here we show that the usage of additional and different motif repeats proves effective in predicting infection. Furthermore, we use machine learning model explanability methods to highlight which of the repeat types are most important, thereby suggesting potential targets for future versions of rapid diagnostic tests.
-
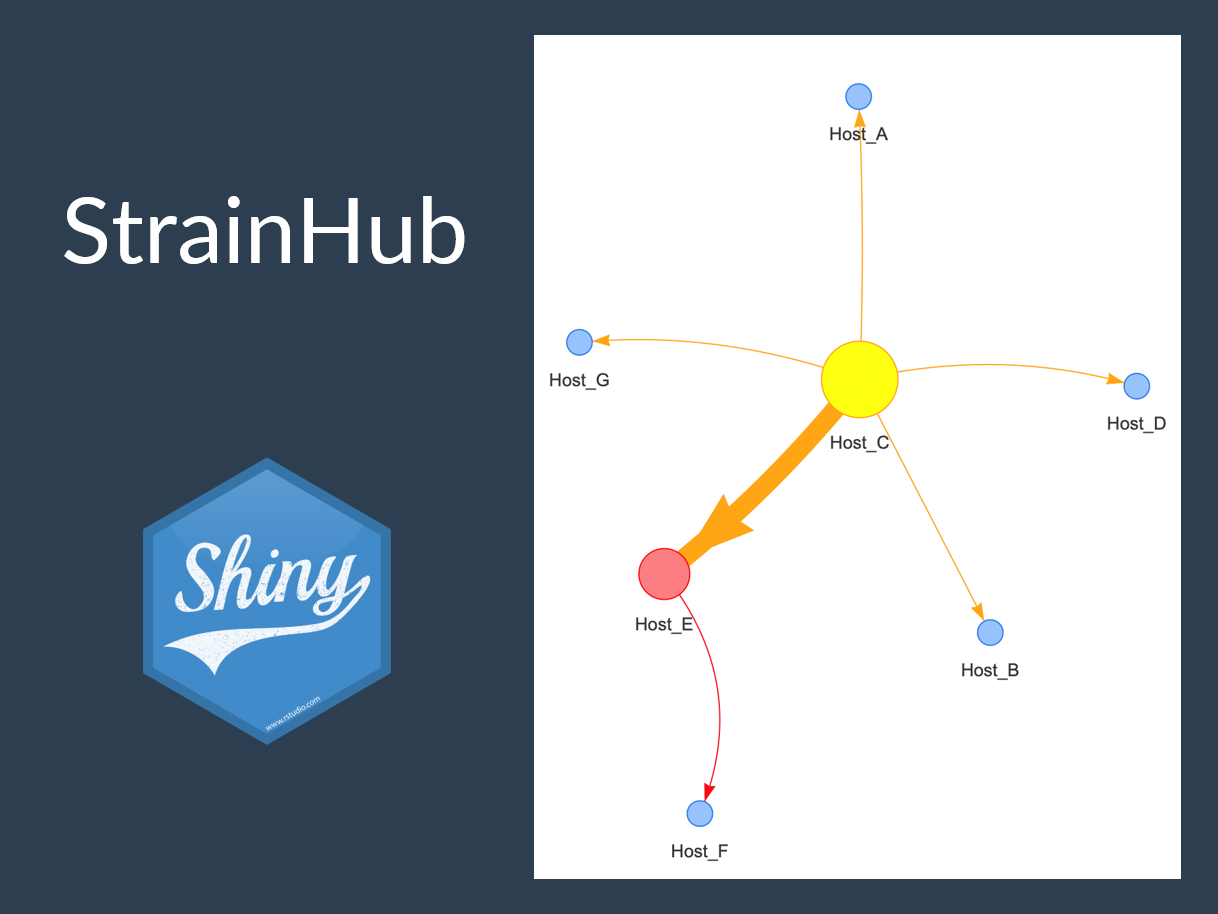
Genomics, Phylogenetics, Infectious Diseases
StrainHub
Shiny web application for visualizing disease transmittion networks from phylogenetic trees.
Strainhub is designed as a web-based software to generate disease transmission networks and associated metrics from a combination of a phylogenetic tree and a metadata associated file. The software maps the metadata onto the tree and performs a parsimony ancestry reconstruction step to create links between the associated metadata and enable the construction of the network.
-

Genomics, Infectious Diseases
Genetic Capitalism in E. coli
Persistence of Genes that Confer Antimicrobial Resistance in the History of Escherichia coli Genomes
Antimicrobial resistance (AMR) in pathogenic strains of bacteria, such as Escherichia coli (E. coli), adversely impact personal and public health. In this study, we examine competing hypotheses for the evolution of AMR including: 1) "genetic capitalism" in which genotypes that confer antibiotic resistance are gained and not often lost in lineages, and 2) "stabilizing selection" in which genotypes that confer antibiotic resistance are gained and lost often. To test these hypotheses, we assembled a dataset that includes annotations for 409 AMR genotypes and a phylogenetic tree based on genome-wide single nucleotide polymorphisms from 29,255 isolates of E. coli. We used phylogenetic methods to count the times each AMR genotype was gained and lost across the tree and used model-based clustering of the genotypes with respect to their gain and loss rates. We demonstrate that many genotypes cluster to support the hypothesis for genetic capitalism while a few cluster to support the hypothesis for stabilizing selection. Comparing the sets of genotypes that fall under each of the hypotheses, we found a statistically significant difference in the breakdown of resistance mechanisms through which the AMR genotypes function. The result that many AMR genotypes cluster under genetic capitalism reflects that strong positive selective forces, primarily induced by human industrialization of antibiotics, outweigh the potential fitness costs to the bacterial lineages for carrying the AMR genotypes. We expect genetic capitalism to further drive bacterial lineages to resist antibiotics. We find that antibiotics that function via replacement and efflux tend to behave under stabilizing selection and thus may be valuable in an antibiotic cycling strategy.
-

R Package, Genomics
msgen
R functions for interfacing with the Microsoft Genomics service in Azure.
The Microsoft Genomics service in Azure can power genome sequencing using a cloud implementation of the Burrows-Wheeler Aligner (BWA) and the Genome Analysis Toolkit (GATK) for secondary analysis. The pipeline can take in multiple FASTQ and BAM files and provides alignment and variant outputs. The msgen package provides an interface to use the service from within R.
-

R Package, Genomics
parEBEN
Parallel Implementations of the Empirical Bayesian Elastic Net Cross-Validation in R.
The Empirical Bayesian Elastic Net (EBEN) algorithm was developed by Huang et al. for handling multicollinearity in generalized linear regression models. Historically, this has been used in the analysis of quantitative trait loci (QTLs) and gene-gene interactions (epistasis). In addition to the algorithm, the group also created the EBEN package for R. This package includes functions to generate the elastic nets for both binomial and gaussian priors. These functions are efficient and do not require large amounts of computational time. However, the package also includes functions for the cross-validation of those models. While essential, this step is a considerably more complex task. The cross-validation functions perform a sweep to determine hyperparameters and minimize prediction error. More specifically, an n-fold cross-validation sweep is performed to minimize error by trying combinations of two parameters (α and λ) in a stepped manner. Experimentally, it has been shown that this can take a rather extended amount of time, especially on larger datasets (as seen in genomics problems).


-

Human Genomics, Machine Learning
MPS-IIIB/NAGLU Prediction
PolyPhen2 + Machine Learning prediction of the effects of genetic mutations on mucopolysaccharidosis IIIB.







Let's Connect!
Want to collaborate, connect, or a consult? Let's chat and see how we can work together to do impactful things...